10.6 微生物单组学快速分析示例
本章节将利用示例数据,完成一个微生物单组学的从导入的完成基本常见的分析流程。
示例数据下载:1. Github下载地址 2. 百度网盘下载地址
10.6.1 微生物数据导入
由于单一组学项目不涉及创建关系表,因此表达矩阵的样本名称必须与表型数据中样本名称完全一致。
library(EasyMultiProfiler)
meta_data <- read.table('coldata.txt',header = T,row.names = 1)
data <- read.table('tax.txt',header = T,sep = '\t')
MAE <- EMP_easy_import(data = data,coldata = meta_data,type = 'tax')
10.6.2 微生物数据查看
查看当前微生物组学
MAE |>
EMP_assay_extract() # 查看表达矩阵
MAE |>
EMP_coldata_extract() # 查看表型数据
MAE |>
EMP_rowdata_extract() # 查看物种注释
查看当前微生物组学种级别数据
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Species')
查看当前微生物组学纲级别数据
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Class')
查看当前微生物组学门级别数据
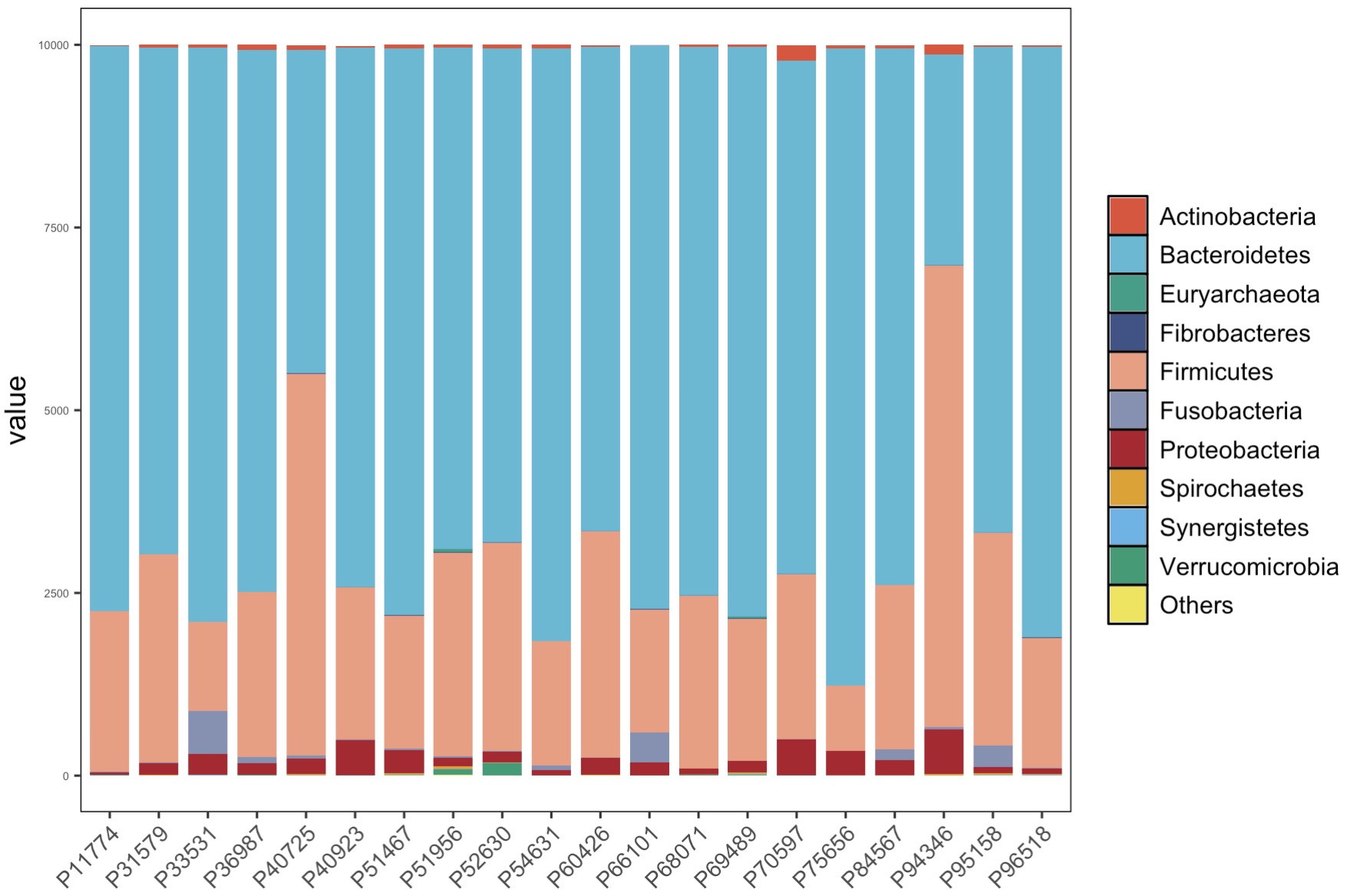
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Phylum') |>
EMP_structure_plot(top_num = 10)
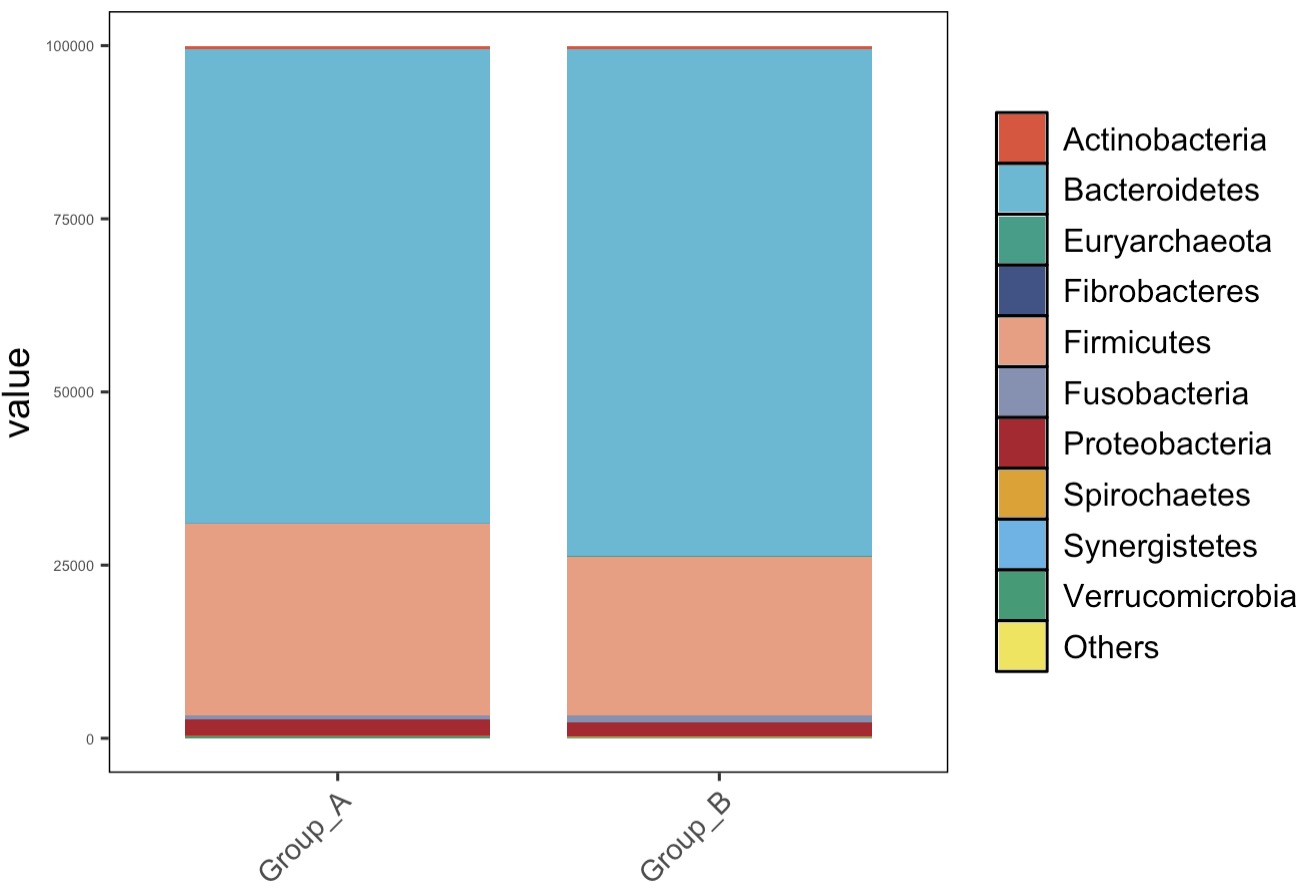
查看当前微生物组学门级别数据并按照组进行折叠
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Phylum') |>
EMP_collapse(collapse_by = 'col',estimate_group = 'Group') |>
EMP_structure_plot(top_num = 10)
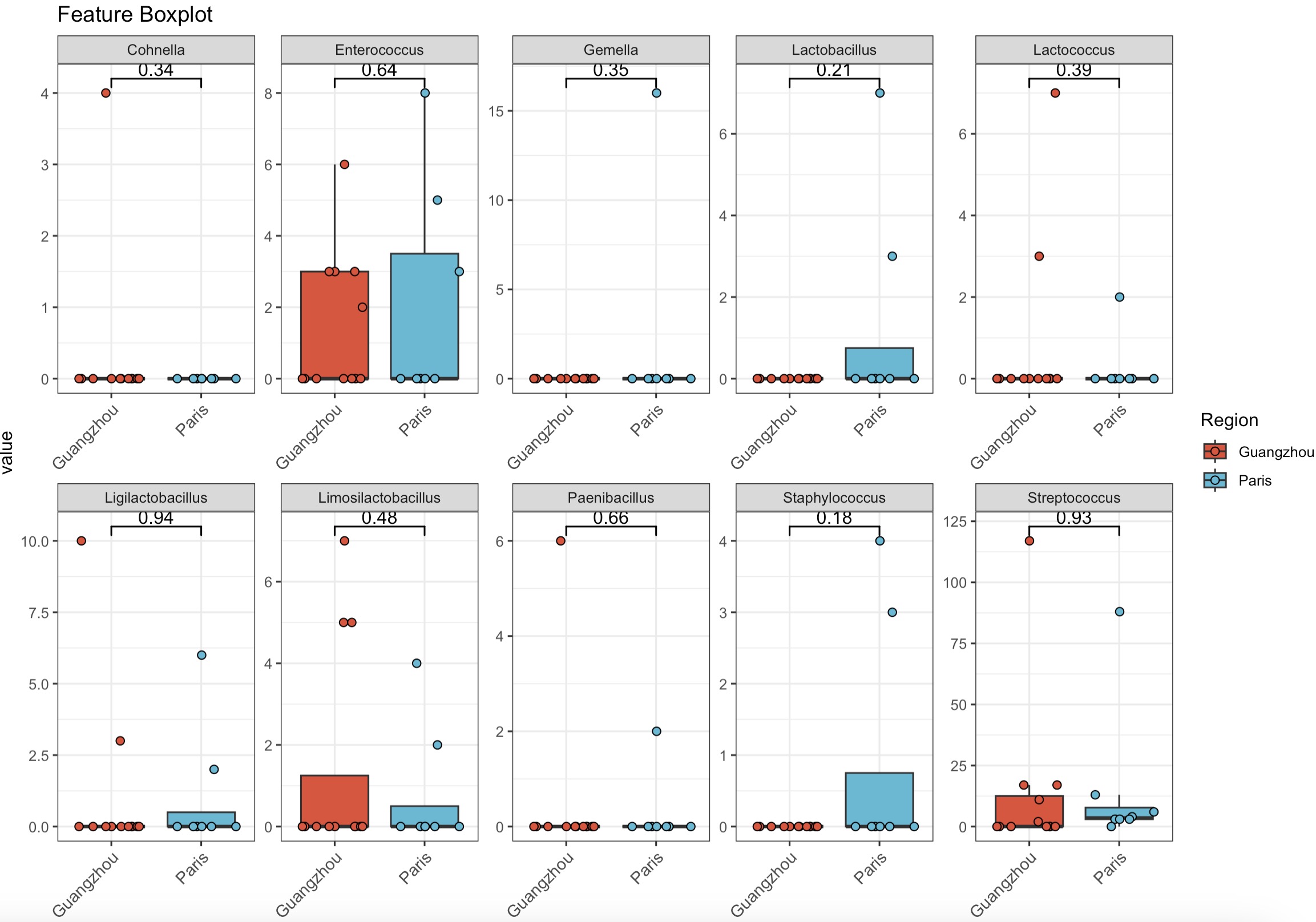
查看Bacilli纲级别下的菌属,并按照区域分组进行统计学检验
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_filter(feature_condition = Class %in% 'Bacilli') |>
EMP_boxplot(estimate_group='Region',
method='t.test',
ncol=5)
10.6.3 微生物数据抽平 (非必须)
按照当前样本中最小读数进行抽平
MAE |>
EMP_assay_extract() |>
EMP_rrarefy()
自定义最小读数进行抽平
MAE |>
EMP_assay_extract() |>
EMP_rrarefy(raresize = 5000)
10.6.4 微生物数据标准化
在属水平上转化成相对丰度
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_decostand(method = 'relative')
在属水平上进行clr转化
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_decostand(method = 'clr')
在属水平上进行对数转化
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_decostand(method = 'log2+1')
10.6.5 微生物的批次矫正 (非必须)
根据表型数据中的区域因素,在属水平上进行批次矫正
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_adjust_abundance(.factor_unwanted = 'Region',
.factor_of_interest = 'Group',
method = 'combat_seq')
10.6.6 微生物核心菌种筛选
筛选出最小丰度0.001,且在至少一个分组的样本占有率超过70%的核心属
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_identify_assay(estimate_group = 'Group',method = 'default',
min=0.001,min_ratio = 0.7)
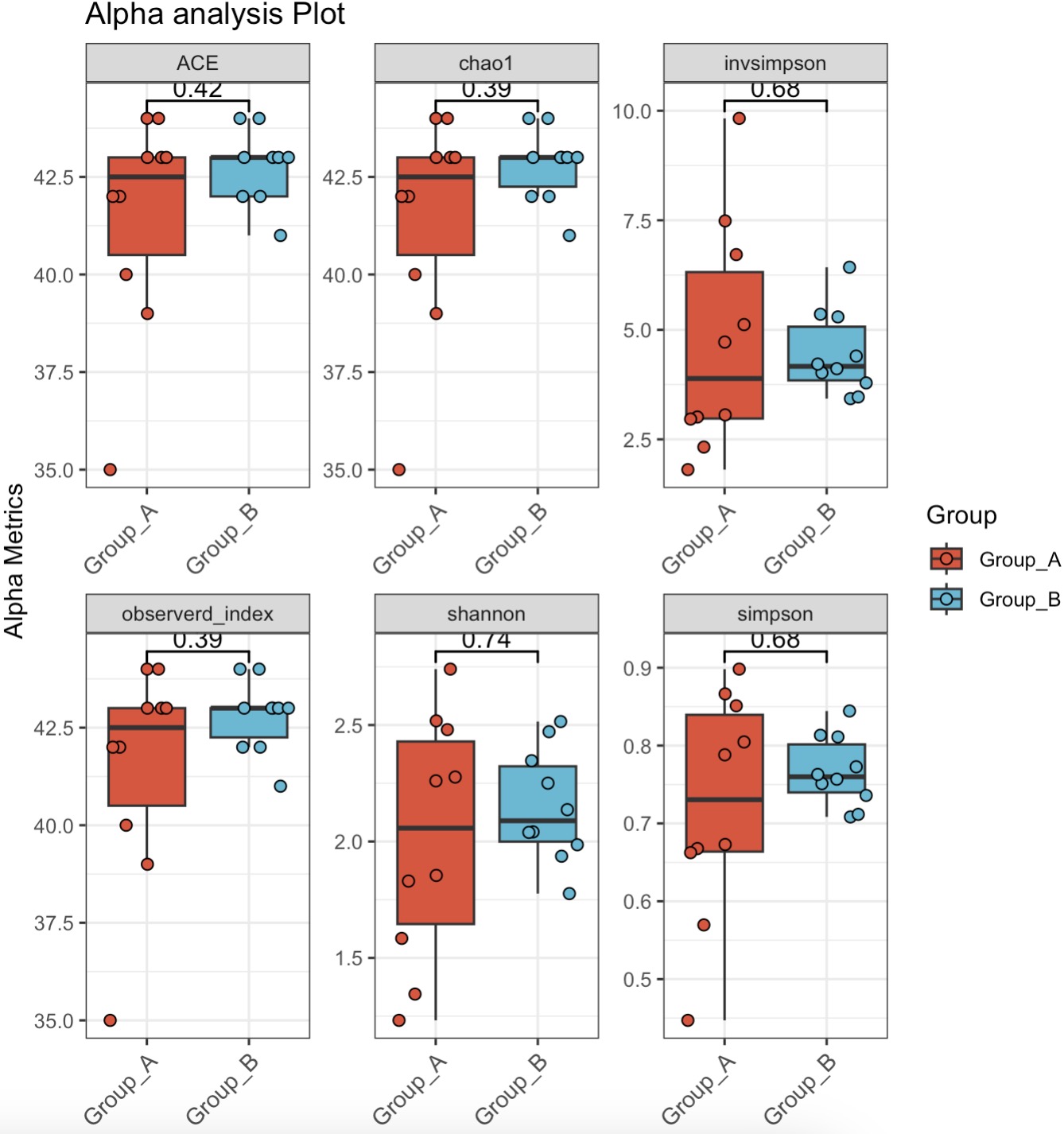
10.6.7 微生物alpha多样性分析
计算核心属的alpha多样性并绘制成箱型图
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_identify_assay(estimate_group = 'Group',method = 'default',
min=0.001,min_ratio = 0.7) |>
EMP_alpha_analysis() |>
EMP_boxplot(estimate_group = 'Group')
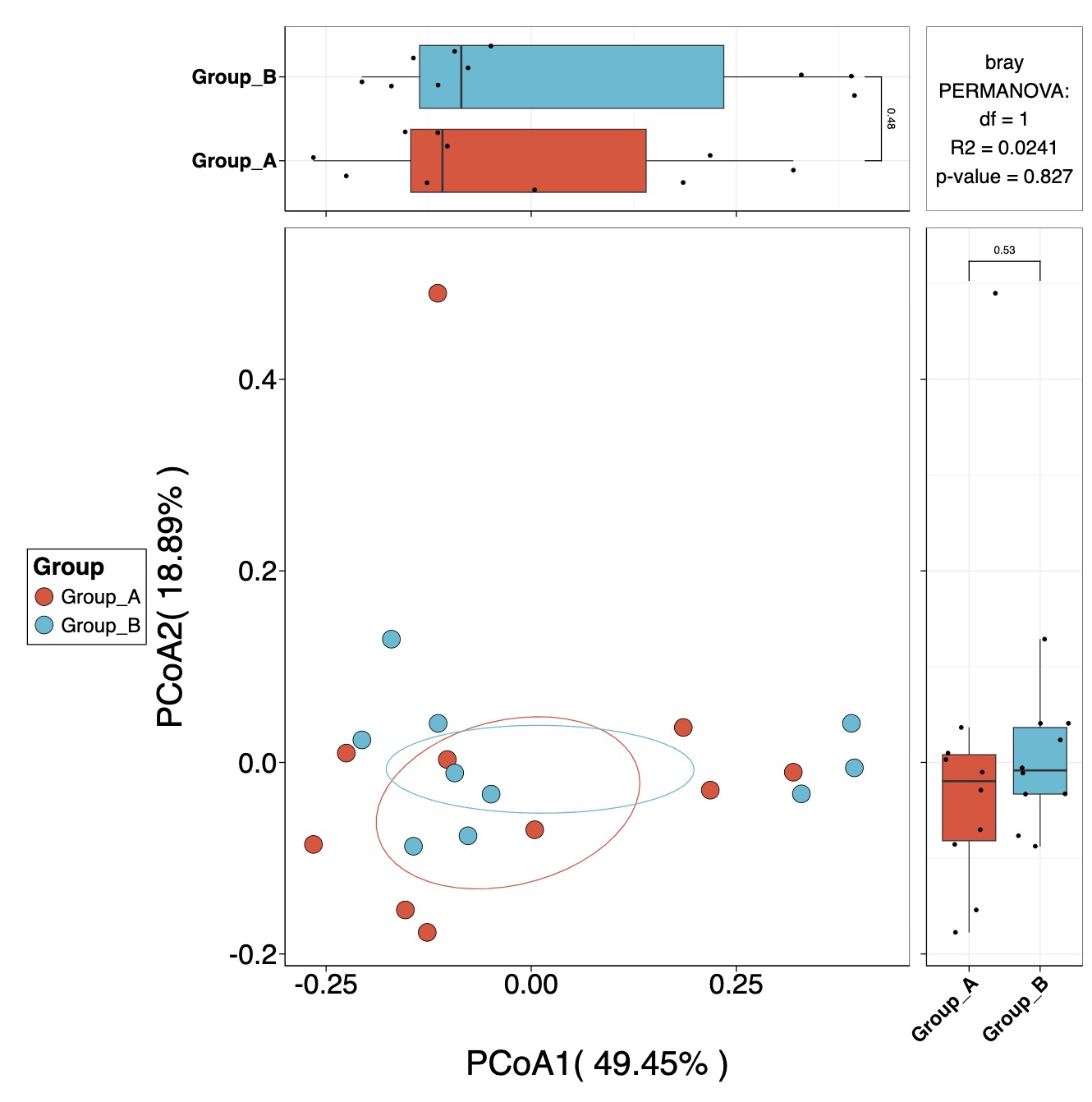
10.6.8 微生物beta多样性分析
计算核心属的beta多样性并绘制成图
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_identify_assay(estimate_group = 'Group',method = 'default',
min=0.001,min_ratio = 0.7) |>
EMP_dimension_analysis(method = 'pcoa',distance = 'bray') |>
EMP_scatterplot(estimate_group = 'Group',show='p12html',ellipse=0.3)
10.6.9 微生物差异分析
计算核心属的wilcox差异性分析,并筛选出差异菌属
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_diff_analysis(method = 'wilcox.test',estimate_group = 'Group') |>
EMP_filter(feature_condition = pvalue < 0.05)
计算核心属的DESeq2差异性分析,并筛选出差异菌属
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group) |>
EMP_filter(feature_condition = pvalue < 0.05,keep_result = TRUE)
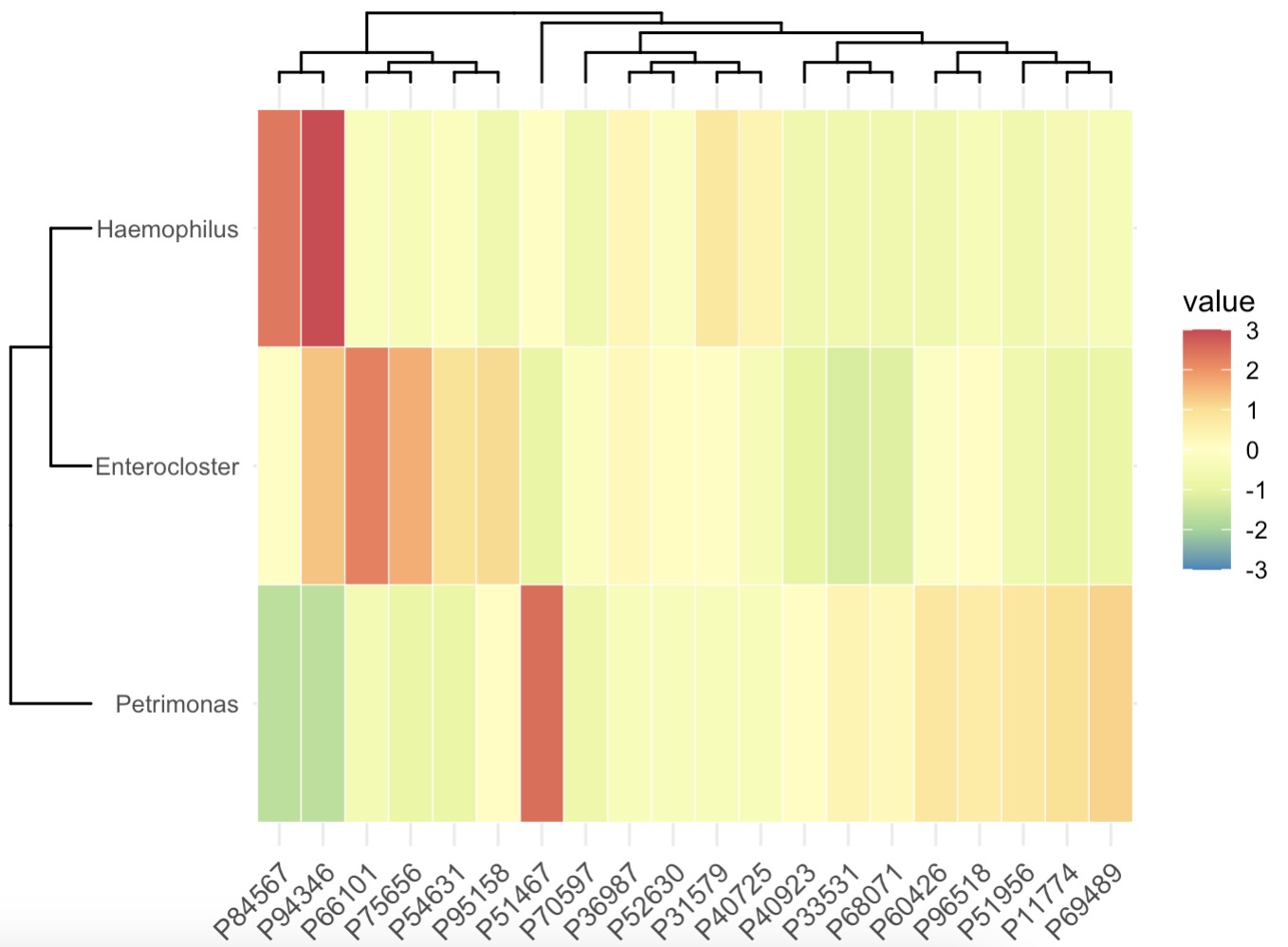
10.6.10 微生物机器学习关键菌
EMP包内置了Boruta算法、随机森林算法、xgboost算法和Lasso算法进行特征筛选。 详细用法可以使用help(EMP_marker_analysis)查看更多示例。
使用Boruta算法筛选出潜在有价值的菌属并绘制成热图
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_identify_assay(estimate_group = 'Group',method = 'default',
min=0.001,min_ratio = 0.7) |>
EMP_marker_analysis(method = 'boruta',estimate_group = 'Group') |>
EMP_filter(feature_condition = Boruta_decision!= 'Rejected') |>
EMP_heatmap_plot(palette='Spectral',legend_bar='auto',
scale='standardize',
clust_row=TRUE,clust_col=TRUE)
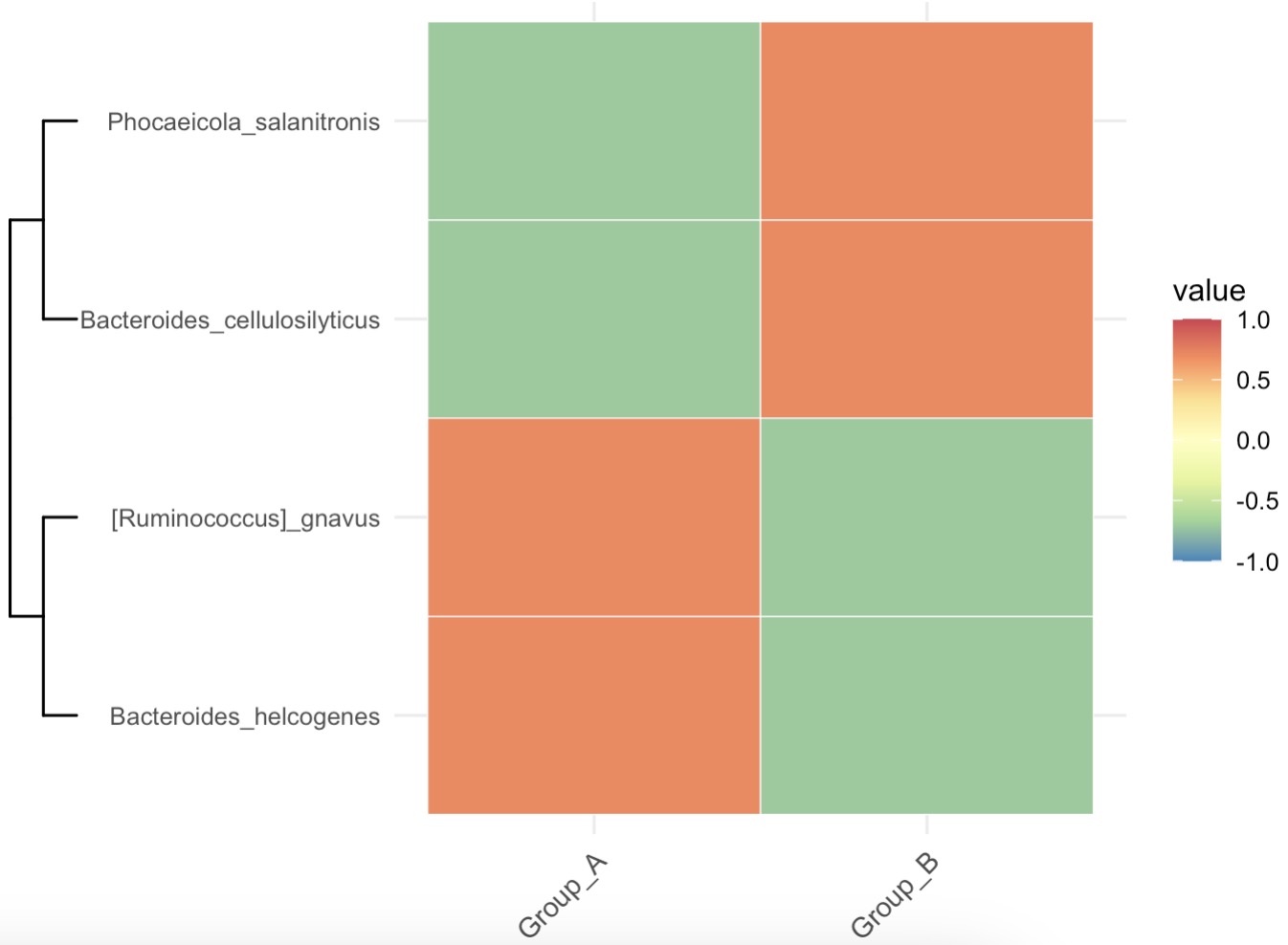
使用Lasso算法筛选出与表型数据中Height密切相关的菌属并按照分组绘制成热图
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Species') |>
EMP_identify_assay(estimate_group = 'Group',method = 'default',
min=0.001,min_ratio = 0.7) |>
EMP_marker_analysis(method = 'lasso',estimate_group = 'Height') |>
EMP_filter(feature_condition = lasso_coe > 0) |>
EMP_collapse(method = 'mean',estimate_group = 'Group',
collapse_by = 'col') |>
EMP_heatmap_plot(palette='Spectral',legend_bar='auto',
clust_row = TRUE,
scale='standardize')
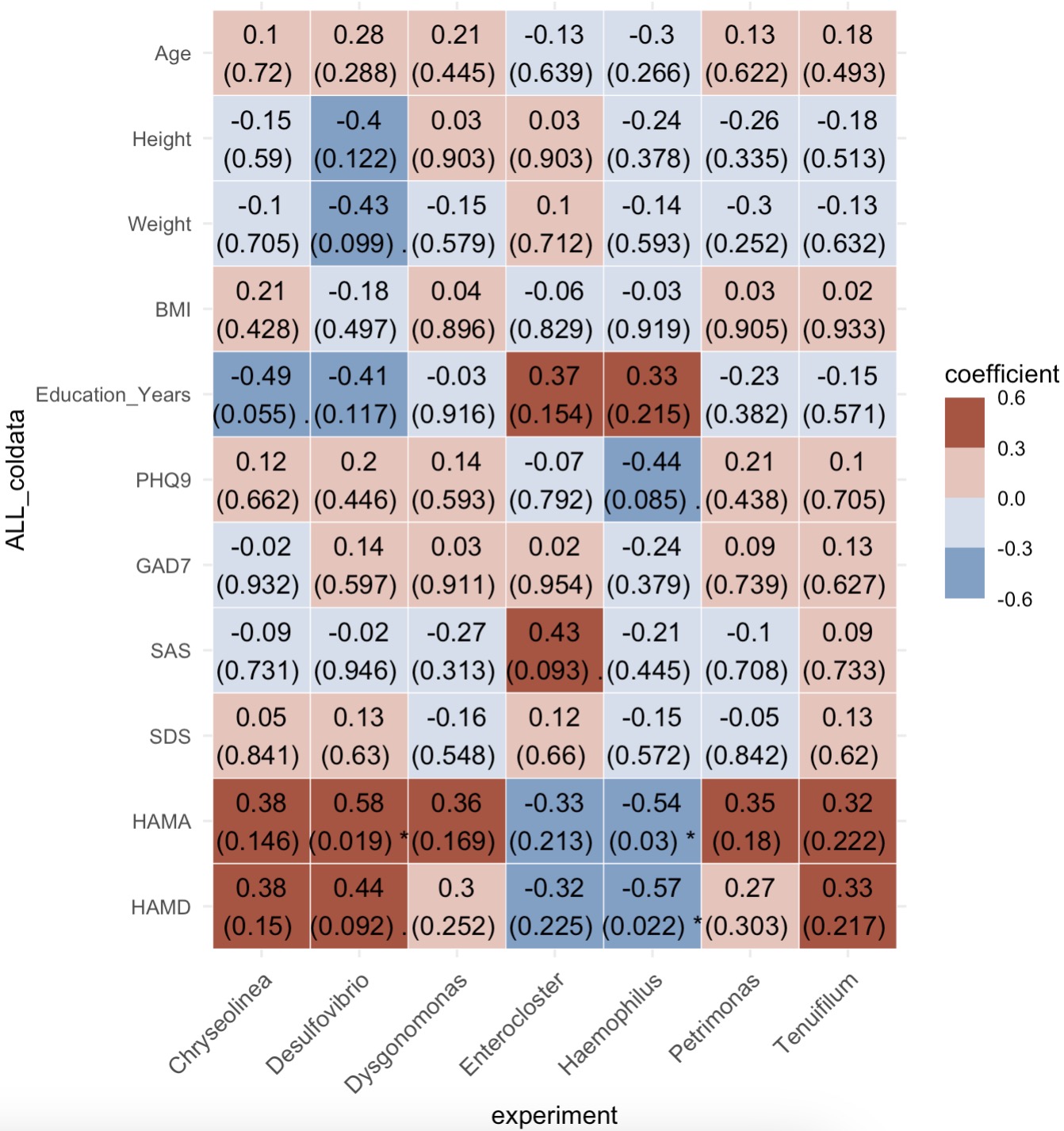
10.6.11 微生物与表型的相关性分析
绘制差异菌属与表型数据相关性热图
diff_genus <- MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_diff_analysis(method = 'wilcox.test',estimate_group = 'Group') |>
EMP_filter(feature_condition = pvalue < 0.05)
meta_data <- MAE |>
EMP_coldata_extract(action = 'add')
(phylum_data + meta_data) |>
EMP_cor_analysis() |>
EMP_heatmap_plot()
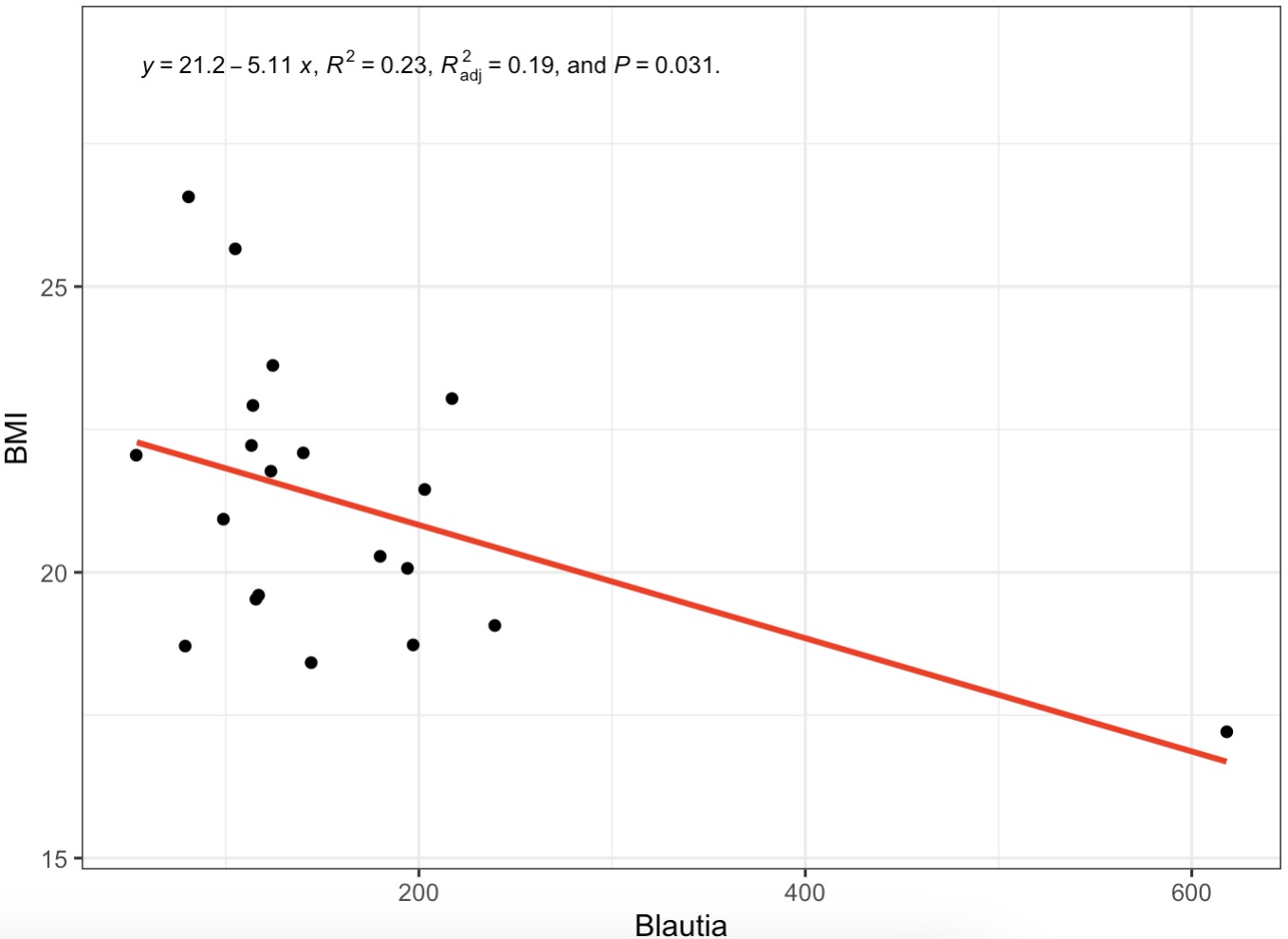
10.6.12 微生物与表型的线性拟合
绘制Blautia菌属与表型数据中BMI的线性拟合
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_fitline_plot(var_select=c('Blautia','BMI'))
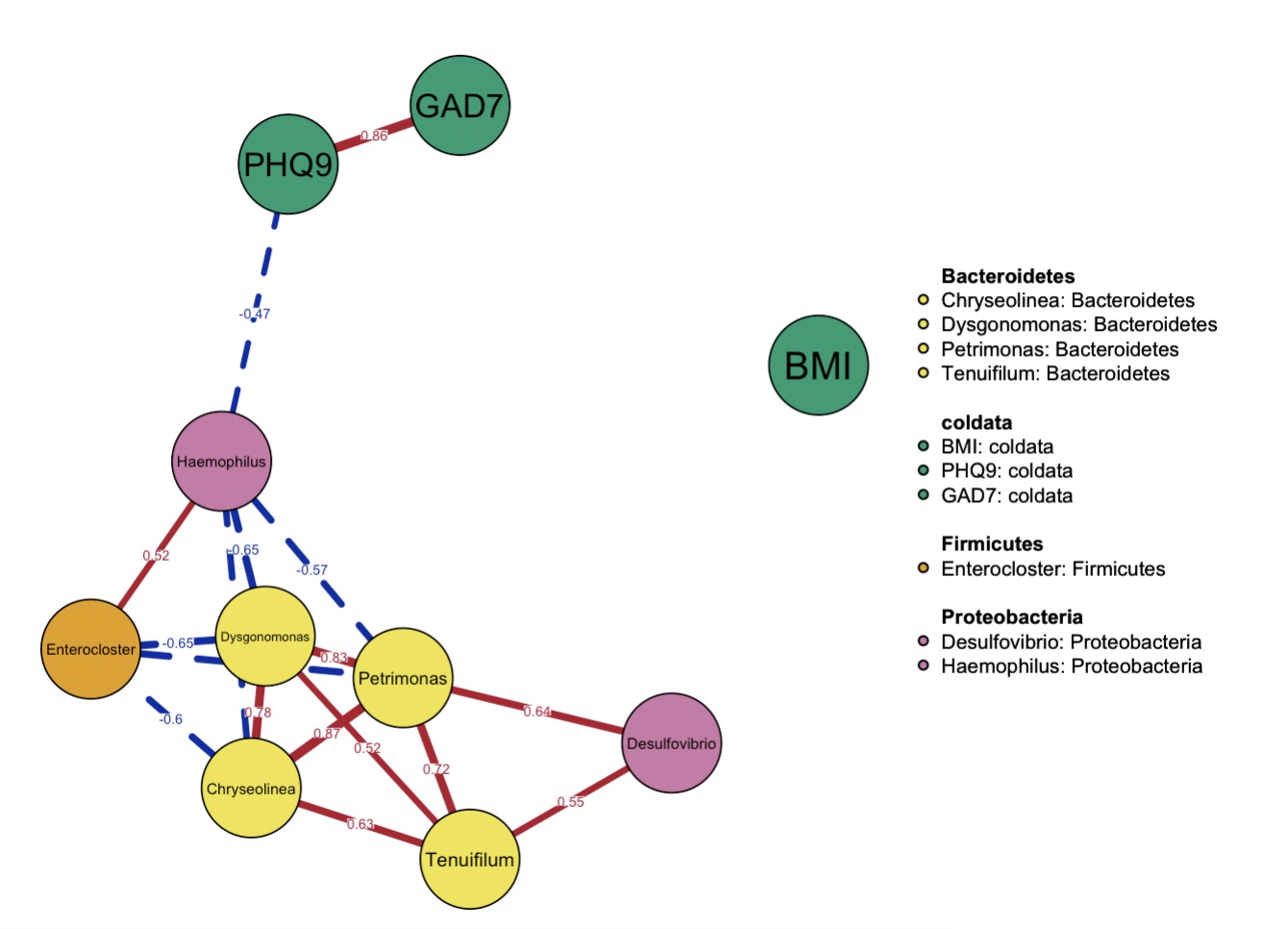
10.6.13 微生物的网络分析
筛选出差异菌属并与部分表型数据绘制网络分析图
MAE |>
EMP_assay_extract() |>
EMP_collapse(estimate_group = 'Genus',collapse_by = 'row') |>
EMP_diff_analysis(method='wilcox.test', estimate_group = 'Group') |>
EMP_filter(feature_condition = pvalue<0.05) |>
EMP_network_analysis(coldata_to_assay = c('BMI','PHQ9','GAD7')) |>
EMP_network_plot(node_info = 'Phylum',label.cex = 1,edge.labels = TRUE)
10.6.14 微生物的mantel分析及可视化
🏷️示例:利用BMI计算差异菌属和双歧杆菌的微生物集与表型数据进行mantel分析
第一步:安装并加载linkET包
devtools::install_github("Hy4m/linkET")
library(linkET)
第二步:利用BMI形成新的分组,并使用oneway差异分析筛选显著差异的菌属
diff_genus <- MAE |>
EMP_assay_extract() |>
EMP_mutate(Degree = dplyr::case_when(
BMI < 18.5 ~ "Lean",
BMI >= 18.5 & BMI < 24 ~ "Normal",
BMI >= 24 & BMI < 28 ~ "Fat",
TRUE ~ "Need Med"
),mutate_by = 'sample',location = 'coldata',.after = Group)|>
EMP_decostand(method = 'relative') |>
EMP_collapse(estimate_group = 'Genus',collapse_by = 'row') |>
EMP_diff_analysis(method = 'oneway.test',estimate_group = 'Degree') |>
EMP_filter(feature_condition = pvalue < 0.05) |>
EMP_assay_extract(action = 'get')
diff_genus
第三步:筛选双歧杆菌的菌属
Bifidobacterium <- MAE2 |>
EMP_assay_extract() |>
EMP_decostand(method = 'relative') |>
EMP_assay_extract(pattern = 'Bifidobacterium',
pattern_ref = 'Genus',action = 'get')
Bifidobacterium
第四步:合并两个微生物数据集并提取出表型数据
tax_data <- purrr::reduce(list(Bifidobacterium,diff_genus),
inner_join,by='primary') |>
tibble::column_to_rownames('primary')
meta_data <- MAE |>
EMP_coldata_extract(action = 'get') |>
dplyr::select(primary,Weight,BMI,Height,Education_Years) |>
tibble::column_to_rownames('primary')
第五步:构建微生物数据和表型数据的mantel分析
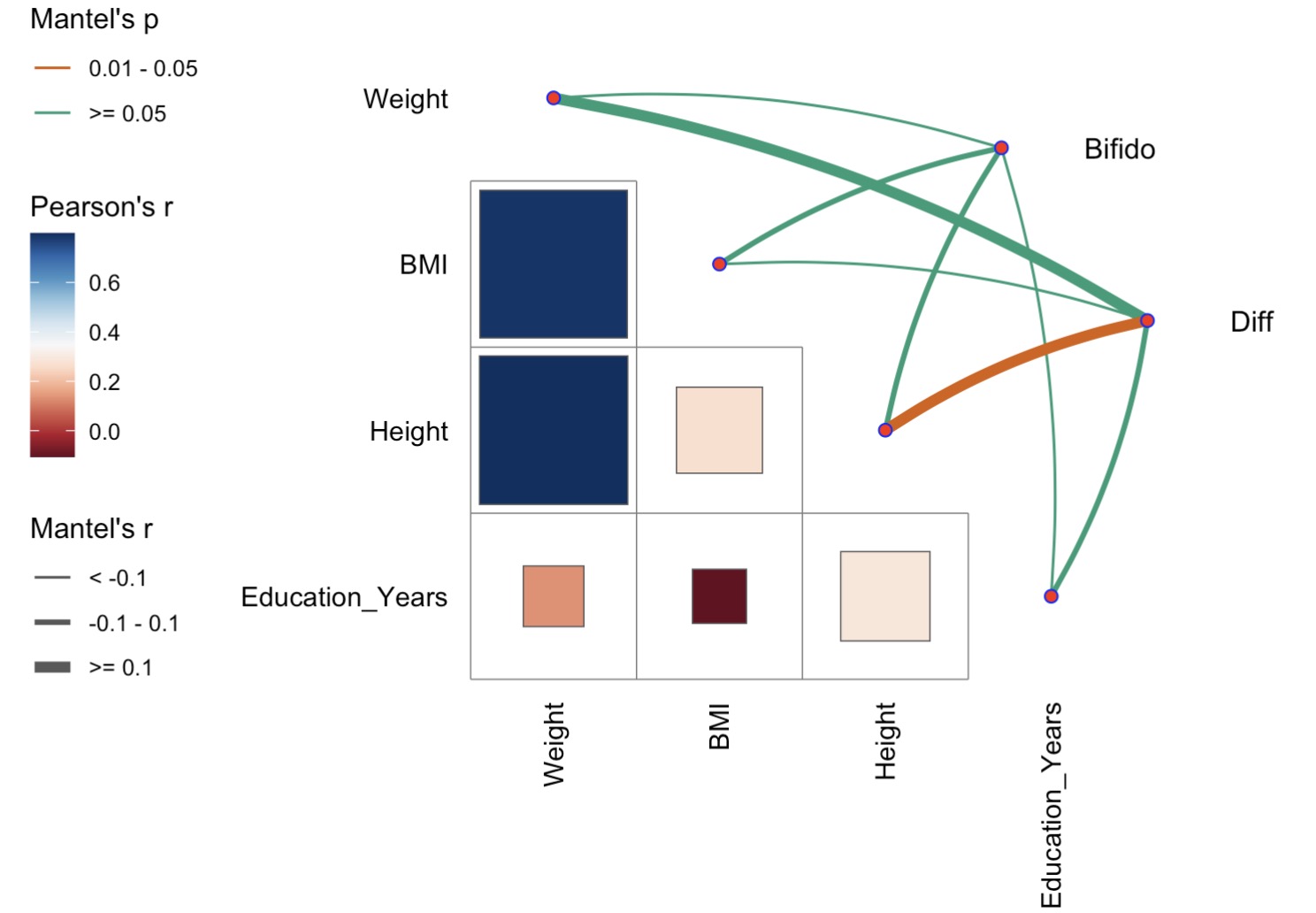
mantel <- mantel_test(tax_data, meta_data,
spec_select = list(Bifido = 1:7,
Diff = 8:10)) |>
mutate(rd = cut(r, breaks = c(-Inf, -0.1, 0.1, Inf),
labels = c("< -0.1", "-0.1 - 0.1", ">= 0.1")),
pd = cut(p, breaks = c(-Inf, 0.01, 0.05, Inf),
labels = c("< 0.01", "0.01 - 0.05", ">= 0.05")))
mantel
第六步:绘制mantel可视化结果
qcorrplot(correlate(meta_data,method='pearson'), type = "lower", diag = FALSE) +
geom_square() +
geom_couple(aes(colour = pd, size = rd),
data = mantel,
curvature = nice_curvature()) +
scale_fill_gradientn(colours = RColorBrewer::brewer.pal(11, "RdBu")) +
scale_size_manual(values = c(0.5, 1, 2)) +
scale_colour_manual(values = color_pal(3)) +
guides(size = guide_legend(title = "Mantel's r",
override.aes = list(colour = "grey35"),
order = 2),
colour = guide_legend(title = "Mantel's p",
override.aes = list(size = 2),
order = 1),
fill = guide_colorbar(title = "Pearson's r", order = 2))